
表的内容
- 公共数据库中的DNA和蛋白质序列的数量非常大。
- 在数据库中搜索涉及将查询序列对齐到数据库中的每个序列,以查找大量的本地对齐。
- BLAST和FASTA是两个相似性搜索程序,其基于过量的序列相似性识别同源DNA序列和蛋白质。
- 它们提供与现有DNA和蛋白质数据库比较DNA和蛋白质序列的设施。
- 他们是两个主要启发式算法用于执行数据库搜索。
Fasta和Blast的工作
- Fasta和Blast是生物信息学中使用的软件工具。BLAST和FASTA都使用启发式字法进行快速成对序列对齐。
- 它通过在两个序列中找到短延伸的相同或几乎相同的字母。这些短串字符称为单词。
- 基本假设是两个相关序列必须至少有一个共同词。
- 通过首先识别单词匹配,可以通过从单词扩展相似区域来获得更长的对齐。
- 一旦找到序列相似性高的区域,相邻的得分高的区域可以加入到一个完整的对齐。
Blast和Fasta之间的主要区别在于,爆炸主要涉及发现未拍摄的局部最佳序列比对,而Fasta参与在较差相似的序列之间的查找相似之处。
BLAST(基本局部对齐搜索工具)
- 爆炸计划于1990年由NCBI斯蒂芬阿尔科尔开发,自成立以来成为最受欢迎的序列分析计划之一。
- BLAST使用启发式对准数据库中的所有序列来对齐查询序列。
- 目的是在相关序列中找到得分高的未搭接片段。在给定阈值以上的片段的存在表明了非随机的两两相似,这有助于从数据库中区分相关序列和不相关序列。
- BLAST是一种流行的生物信息学工具,因为它能够快速识别两个序列之间的局部相似性区域。BLAST计算一个期望值,该期望值估计两个序列之间的匹配数。它使用序列的局部对齐。
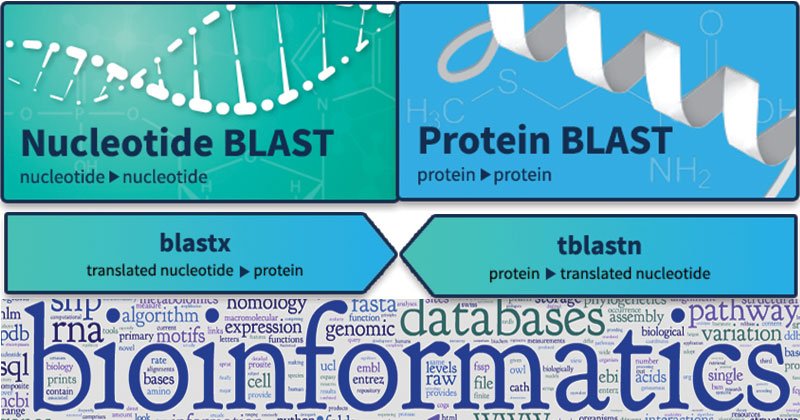
爆炸的变体
- BLAST-N:将核苷酸序列与核苷酸序列进行比较
- BLAST-P:将蛋白质序列与蛋白质序列进行比较
- BLAST-X:将核苷酸序列与蛋白质序列进行比较
- TBLAST-N.:将蛋白质序列与核苷酸序列的六帧翻译进行比较
- tblast-x:比较核苷酸序列对蛋白序列六帧翻译的六帧翻译。
Fasta.
- FASTA是fast-all的缩写。
- 它是先于BLAST开发的第一个数据库相似度搜索工具。
- FASTA是另一个序列对准工具,用于搜索DNA和蛋白序列之间的相似性。
- Fasta使用“散列”策略来查找匹配的匹配短延伸的相同残留物,长度为k。残留串被称为ktuples或ktups,其等同于爆炸中的单词,但通常比单词短。
- 通常,Ktup由两个用于蛋白质序列的残基和DNA序列的六个残基组成。
- 因此,查询序列被分解为序列模式或称为k元组的单词,目标序列将搜索这些k元组,以找到两者之间的相似性。
- Fasta是一个有关类似搜索的精细工具。
这些方法不保证找到最佳的对齐或真实同源物,但比动态编程快50-100倍。
参考文献
- 熊j .(2006)。重要的生物信息学。德州农工大学。剑桥大学出版社。
- Arthur M Lesk(2014)。生物信息学介绍。牛津大学出版社。牛津,英国
- http://pediaa.com/difference-between-blast-and-fasta/#Blast.
- https://embnet.vital-it.ch/CoursEMBnet/Basel03/slides/BLAST_FASTA.pdf
- https://www.slideshare.net / aavrilcoghlan/blast-16572940
- https://www.cs.helsinki.fi/bioinformatiikka/mbi/courses/0708/itb/slides/itb0708_slides_83-116.pdf.
- https://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs&DOC_TYPE=BlastHelp