
表的内容
引物是什么?
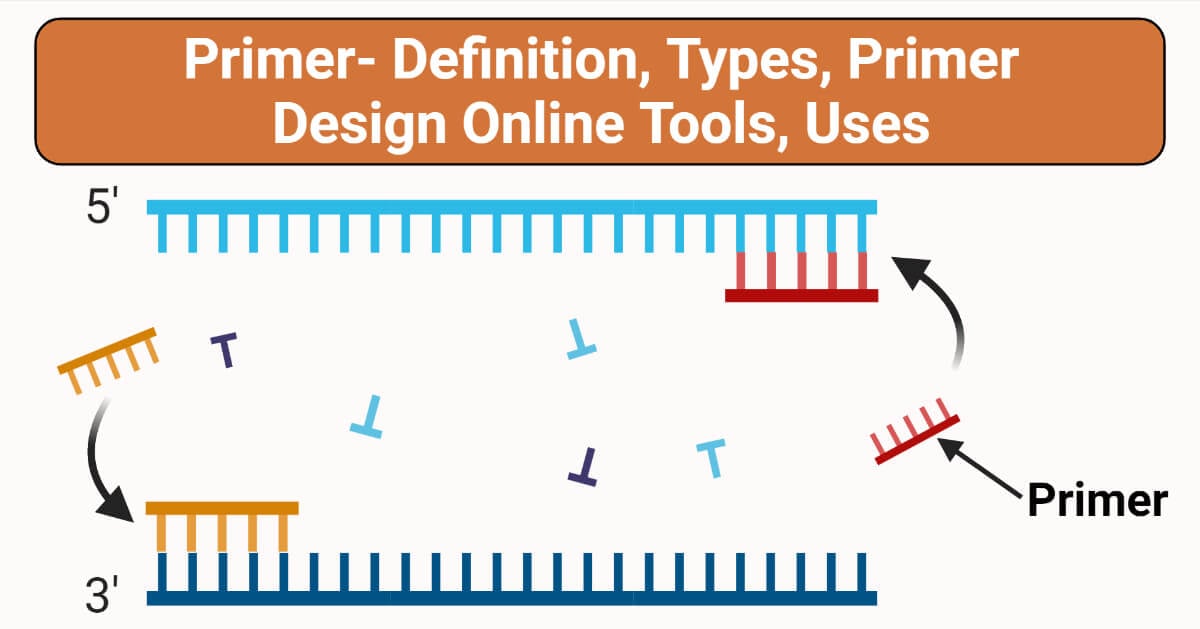
引物是作为DNA合成起始点的一小段序列。可以有一组与模板DNA序列互补的引物(正向和反向)-起始合成点。
引物的主要目的是合成具有自由末端和聚合酶起始点的DNA。一对引物,一个在模板链上,另一个在互补链上,在被设计的序列的两端结合,同样,3 '对应模板链进行延伸。
正向引物为3 ' -5 ',反向引物为5 ' -3 '。然而,延伸过程产生了两条新的ds DNA链。
注意:当模板链与互补链位于一起时,二聚体的形成是不利的条件。
正向引物和反向引物不遵循互补规则,而是正向引物与靶标5 ' P的一端结合,而5 ' P的另一端则占用反向引物。
DNA引物的应用最为广泛聚合酶链反应,不像复制,由于:
- 稳定性的概念在DNA引物中比RNA引物多。
- 由于是单向聚合方式,反应结束后RNA引物无法去除。
- DNA引物很容易合成
- 然而,大多数扩增是通过DNA进行的,建议只使用特定的核苷酸引物。
注:引物参与复制过程,在复制结束时能够去除RNA引物,而DNA引物则不能。
引物类型(DNA引物vs RNA引物)
单独的活生物体使用RNA引物,而体外则使用DNA引物。然而,由于多种原因,如稳定性、易于储存、启动合成所需的酶较少等,DNA引物更受青睐。
之间的比较DNA和RNA下面列出了基础知识。
DNA引物:体外:PCR扩增,DNA测序。
RNA引物:体内:DNA复制、克隆。
反应
DNA引物:扩增过程依赖于温度,蛋白质较少。
RNA引物:该复制过程是一种催化反应,在酶依赖的方式与几种蛋白质。
长度
DNA引物:18- 24个碱基对。
RNA引物:10- 20个碱基对。
合成:DNA引物是化学合成的,而RNA引物则需要Primase酶。
DNA引物寿命长且更稳定,而RNA引物寿命短且反应性更强。尽管如此,DNA聚合酶开始向现有核酸活性的3 ' (OH)端添加核苷酸,同时延伸和复制母链。
引物设计
下面,我们来讨论一下Primer设计的基本要素:底漆长度、熔点、底漆退火等参数
引物长度:18-24之间的寡核苷酸被认为是足够安静和有利的,因此短引物在退火温度下很容易与模板结合。
熔化温度(52°C-56°C)该序列的GC结果可以很好地反映引物Tm的存在。但引物的差异不应小于2°C。
引物退火(Ta):过高的Ta会导致引物模板杂交不足的低PCR产物,而过低的Ta会导致大量碱基对错配导致的非特异性PCR产物。
Ta= 0.3*Tm(引物)+0.7(产物)- 14.9,Tm(引物)
底漆熔化温度:
Tm(引物)-测量最不稳定的引物-模板对。
Tm(产物)-用于测量PCR产物的熔化温度。
改进的步骤退火可以使用梯度PCR进行,其中可以设置温度以结合引物。
引物GC含量及夹钳:基因测序引物必须GC含量在40-60%之间,3’端,用2个GC碱基- GC钳夹。然而,随着引物结合的改善和特异性的提高,具有3h键的GC bp比具有2键的AT键更强。
设置限制性内切酶(RE)切位点我们的引物在总切位点的5 '端添加3-5个碱基,这种被称为先导序列的酶可以获得更高的酶切效率。
稳定性:最大的G允许3 '端至少绑定3-5个碱基。然而,稳定的3 '端可以减少错误启动。
注意设计PCR引物
引物可以形成以下几种类型:
- 发夹:引物分子内相互作用形成的3 '端-2kcal/m和内部发夹-3kcal/m的环结构可以耐受。
- 二聚体:两个引物通过分子间相互作用形成DNA的结构。同样,如果两个同源或相同意义的引物之间形成相互作用,称为自二聚体,而相反的引物称为交叉二聚体。
- 重复和运行在一个核苷酸的连续延伸中连续出现二核苷酸被认为是最重要的特性。最大的没有。重复和运行的是4个二核苷酸和4个碱基对。
- 引物-模板交叉同源性:引物的设计应注意模板内除了导致非特异性结合和扩增的目标位点以外的其他同源性。这可以分为两类:a)Intra-primer同源性:在3个以上碱基的区域内,同一对碱基中的互补碱基可以形成分子内键b)Inter-primer同源性:具有互补序列的正向和反向引物负责分子间的键合。
分析引物二聚体的形成是首要的重要注意事项。然而,它涉及到吉布斯自由能的确定,这有助于成为一个。虽然5 '端比3 '端更可靠。
最好的初级在线设计工具
- 初级设计工具(nih.gov)
- Primer3输入
- Primer3Plus(生物信息学。问)
- PrimerQuest -设计qPCR检测| IDT
- PerlPrimer (sourceforge.net)
- 用Oligo引物分析软件设计引物
- Real-Time PCR引物设计- Real-Time PCR探针设计- GenScript
- www.autoprime.de
初步设计方案/步骤/过程
引物设计的金门克隆法
这种方法要求使用一个载体组装(质粒)到一个单一的构建具有一个或多个DNA片段。PCR引物与相邻的DNA片段重叠形成限制性位点,设计2S型酶,与片段的DNA连接酶定向组装。同样,这种方法利用了第2类限制位点的使用,即通过非回文黏端悬挑切断它们的限制位点之外。该方法利用多个DNA片段,利用插入片段上的悬挑序列组合,便于与相邻片段进行退火。然而,2S型限制位点允许金门组装,因此克隆后没有限制位点。
应用Gibson克隆法进行引物设计
该方法依赖重组,尽管限制消化和连接质粒的生成。它允许在线性化的载体两侧各有一个20-40 bp长的末端重叠产生相同的同源序列,用于两个目标DNA片段。它结合了被外切酶切割的完全相同的重叠部分,因此在组装时互补部分伸出到它上面。简而言之,这可以定义为一种方法,认为两个同源DNA片段在一起,重叠的DNA末端与他们融合。
qPCR中引物和探针的验证
利用现有工具设计qPCR引物和探针后,利用BLAST (insilica validation)进行insilica validation,以确认目标基因序列的特异性。BLAST算法对多个数据库使用一组到完整数据库记录的链接缺口比对进行序列相似度搜索(Raymaekers M等人,2009)。查询覆盖率和最大标识应该是100%。然而,BLAST程序报告了一个统计意义,称为每个对齐的“期望值”(E -值),这是一个偶然找到匹配的指标。E -值≤0.01表示同源序列(Altschul等人,1990)(Karlin等人,1990)。评估潜在生物关系的E值测量(Raymaekers M等人,2009)。尽管Insilco工具提供了有价值的反馈,但使用设计的引物和探针进行qPCR检测的特异性必须通过直接的实验证据进行经验验证(Bustin等人,2009)。
qPCR产物的特异性会受到非特异性扩增的影响,可以通过分析熔化曲线(也称为解离曲线)来检查,根据ds DNA结合染料(包括SYBR绿色)生成qPCR协议,因为它们结合引物-二聚体和其他反应伪影产生荧光信号(Holland等,1991年)(Heid CA等,1996年)。熔融曲线在所有已报道的用于扩增后进行qPCR的软件程序中都可以得到(Pfaffl MW, 2004)。
底漆设计的应用
除了PCR, DNA测序引物结合限制性克隆,以及其他DNA新组装方法,如Gibson DNA组装法结合Golden Gate法。
限制
吉布斯自由能在引物设计中起着非常关键的作用。这是因为在恒定的温度和压强下,反应是自发的。因此,较高的G表示(大于0或正的G)有生成的焓,而二级结构的自发反应较低,G值较低。非常负的G值表明结构倾向于形成线性结构,并以反向的方式释放热量,因此应该避免形成更稳定的二级结构(更大的负G值)。
参考文献
- Raymaekers M, Smets R, Maes B等(2009)实时PCR检测方法的优化和验证清单。J临床实验室肛门23:145-151
- Karlin S, Altschul SF(1990)利用一般评分方案评估分子序列特征统计显著性的方法。美国学术期刊A 87:2264-2268
- Altschul SF, Gish W, Miller W等(1990)基本的局部对齐搜索工具。J Mol biool 215:403-410
- Bustin SA, Benes V, Garson JA等人(2009)MIQE指南:发布实时荧光定量PCR实验的最低信息。中国化学55:611 - 622
- Pfaffl MW(2004)实时荧光定量PCR的定量策略。In: Bustin SA (ed) A-Z of Quantitative PCR (IUL Biotechnology, No. 5).国际大学线(IUL),圣地亚哥,CA, pp 87-112
- Holland PM, Abramson RD, Watson R等(1991)利用水生Thermus aquaticus DNA聚合酶的50-30外切酶活性检测特异性聚合酶链反应产物。美国国家科学院学报88:7276-7280
- Heid CA, Stevens J, Livak KJ等(1996)Real time quantitative PCR。基因组Res 6: 986-994