
Database similarity searching is an essential technique in bioinformatics as it allows us to characterize newly determined sequences by comparing them to existingdatabases.
FASTA sim是第一个广泛使用的数据库ilarity search tools. FASTA (or FastA), an abbreviation for ‘Fast-All’, is a sequence alignment tool that takes nucleotide or protein sequences as input and compares it with existing databases.It was first developed by David J. Lipman and William R. Pearson in 1985 and has since been refined and adapted for various applications.
The text-based file format for representing nucleotide or protein sequences, which originates from the FASTA program, has now become a standard in bioinformatics. Many other sequence database search tools also use the FASTA file format.

FASTA Programs
FASTA was originally developed for comparing protein sequences. The original program was referred to as FASTP. It quickly became a popular tool for sequence alignment and database searching. The program has been continually updated and improved. There are now different FASTA programs available, each used for different types of sequence searches:
- FASTAcompares a DNA query sequence against a database of DNA sequences or a protein query sequence against a database of protein sequences using the FASTA algorithm.
- SSEARCHperforms protein-protein or DNA-DNA comparisons using the Smith-Waterman algorithm.
- GGSEARCH/ GLSEARCHworks using a global alignment algorithm (GGSEARCH) or a combination of global and local alignment algorithms (GLSEARCH) to compare protein and nucleotide sequences.
- FASTX/ FASTYcompares a DNA sequence and a database of protein sequences by translating the DNA sequence into three frames and allowing gaps and frameshifts.
- TFASTX/ TFASTYcompares a protein sequence and a database of DNA sequences. The DNA sequence is translated in six frames – three in the forward direction and three in the reverse direction.
- FASTF/ TFASTFcompares mixed peptide sequences against a protein (FASTF) or translated DNA (TFASTF) databases.
- FASTS/ TFASTScompares a set of short peptide fragments against the protein (FASTS) or translated DNA (TFASTS) databases.
How FASTA Works
FASTA works by comparing a query sequence to a database of sequences to identify similar matches. The program uses a heuristic algorithm to quickly search the database and identify the most significant matches.
The working mechanism of FASTA is described in the following steps:
Step 1: Identifying Regions
The first step is identifying regions with high similarity by creating a lookup table for the query sequence. This step is also called hashing step. To create the lookup table, the query sequence is first broken down into smaller words known as k-tuples (ktup).
When the ktup value is increased, the number of background word hits is reduced. By reducing the number of these background word hits, the algorithm can focus on the more relevant hits, enhancing the overall search speed. k-tuple is usually 2 for proteins and 6 for nucleotide sequences.
Once the lookup table is created, it is used to identify matches between the k-tuples in the query sequence and the database sequences. Similar regions are represented as diagonals in a two-dimensional matrix. The ten regions with the highest density of word matches are the high-similarity regions, and these best ten diagonals are saved.
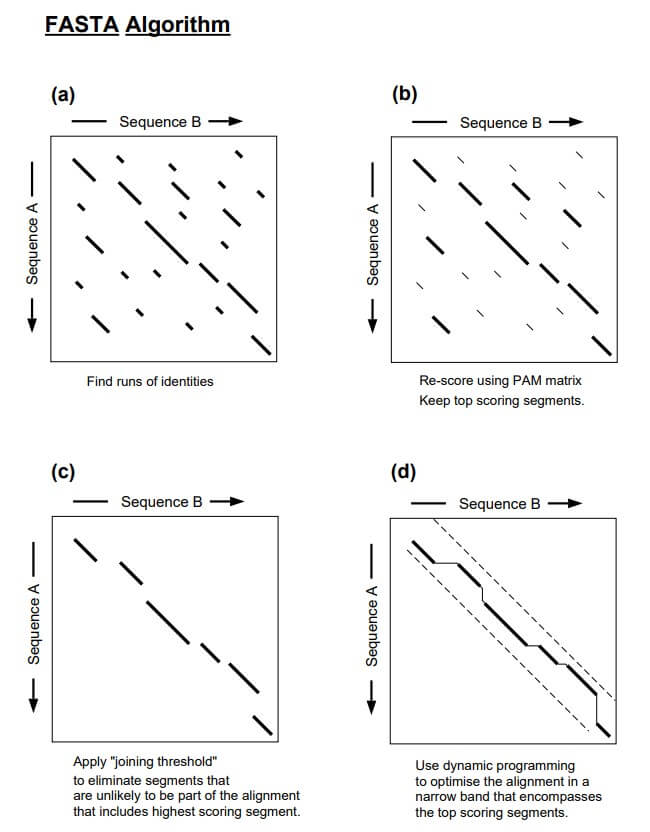
Step 2: Re-Scoring
In the second step, the ten best diagonals are rescored using suitable scoring matrices. For protein, BLOSUM50 or PAM matrix is used; for DNA sequences, the identity matrix is used. A subregion with the highest score is identified for each of the rescanned diagonal regions. These high-scoring subregions within the diagonals are called initial regions.
Step 3: Joining Threshold
Next, a score cutoff or the joining threshold is applied that excludes segments unlikely to be part of the final alignment. The library sequences are ranked based on their initial scores. The regions with initial scores above the pre-set threshold are selected and checked to see if they can be joined together. This step introduces gaps between the diagonals while applying gap penalties. The score of the gapped alignment is calculated by subtracting a penalty for each gap, which is used to rank the database sequences by similarity.
Step 4: Final Alignment
Finally, the gapped alignment is refined to produce the final alignment. This is done by using the banded Smith-Waterman algorithm, which is a dynamic programming algorithm that calculates the optimal score (opt) for alignment. This score is used for statistical calculations.
Statistical Significance and FASTA
FASTA also provides an estimate of the statistical significance of each alignment found. It is evaluated using the E-value, which measures the likelihood of obtaining a sequence alignment score by chance. The smaller the E-value, the more significant the alignment.
E-value is not the only statistical parameter. FASTA also uses other statistical measures, such as the bit score and the similarity score based on the scoring matrix and gap penalties, to evaluate the significance of sequence alignments.
FASTA输出还包括一个额外的statistical parameter, the Z-score, which represents the number of standard deviations from the mean score of the database search. A higher Z-score value indicates a more significant match.
Applications of FASTA
FASTA has a wide range of applications. Some are:
- FASTA can be used in the sequence alignment to identify regions of similarity. This is useful for identifying conserved regions in DNA or protein sequences, which can help to identify functional domains or motifs. Identifying these functional domains or motifs can provide insights into the biological function of the sequence.
- FASTA can be used to search large databases of sequences to find matches to a given query sequence. This helps to identify homologous sequences, which can help to predict the function of a newly identified sequence.
- FASTA can constructphylogenetic treesby aligning sequences from different species and identifying evolutionary relationships between them.
References
- Barton, G. J. (1996). Protein sequence alignment and database scanning. Protein structure prediction: A practical approach, 31-63.
- FASTA Programs (virginia.edu)
- FASTA/SSEARCH/GGSEARCH/GLSEARCH < Sequence Similarity Searching < EMBL-EBI
- Lloyd, Andrew. (2001). Bioinformatics: A Practical Guide to the Analysis of Genes and Proteins (Methods of Biochemical Analysis, 43). Briefings in Bioinformatics. 2. 10.1093/bib/2.4.407.
- Mount, D. W. (2001) Bioinformatics: sequence and genome analysis. Cold Spring Harbor Laboratory Press.
- Pearson, W. R. (2005). FASTA Algorithm. Encyclopedia of Life Sciences. doi:10.1038/npg.els.0005255
- Pearson, W. R. (2016). Finding Protein and Nucleotide Similarities with FASTA. Current Protocols in Bioinformatics, 3.9.1–3.9.25. doi:10.1002/0471250953.bi0309s53
- Pearson, W. R., & Lipman, D. J. (1988). Improved tools for biological sequence comparison. Proceedings of the National Academy of Sciences, 85(8), 2444–2448. doi:10.1073/pnas.85.8.2444
- 桑塞姆,c(2000)。数据库搜索和DNAprotein sequences: An introduction. Briefings in Bioinformatics, 1(1), 22–32. doi:10.1093/bib/1.1.22
- Xiong, J. (2006). Essential Bioinformatics. Cambridge: Cambridge University Press. doi:10.1017/CBO9780511806087